Reducing Missed Calls Rate in Genetic Datasets Made Easy

Reducing missed calls rate (mcr) in genetic datasets plays a vital role in ensuring data quality and research reliability. High rates of missing data can lead to biases, such as errors in identifying SNPs or deviations from Hardy-Weinberg equilibrium. For example, studies often remove markers with low minor allele frequency if their call rate falls below 95% to maintain dataset integrity. Addressing these issues requires actionable strategies. Techniques like Random Forest and kNN have proven effective for handling missing data, outperforming traditional methods in complex scenarios. Tools like Sobot can further enhance data quality by streamlining processes and improving operational efficiency.
Understanding Missed Calls Rate (MCR) in Genetic Datasets
What Is Missed Calls Rate (MCR)?
Definition and examples of missed calls in genetic datasets.
Missed calls rate (MCR) refers to the proportion of missing data in genetic datasets. It measures how often a genetic variant fails to be identified during sequencing. For example, if a dataset contains 1,000 genetic markers and 50 of them lack data, the MCR would be 5%. This metric helps you assess the reliability of your dataset. A high degree of missingness often arises in studies involving complex traits, where sequencing errors or low-quality samples lead to incomplete data.
Imagine analyzing a dataset with a high degree of missingness. You might find gaps in critical regions, making it harder to identify single nucleotide polymorphisms (SNPs). These gaps can distort your findings, especially in studies requiring precise genetic mapping.
Common causes of high missed calls rate (e.g., sequencing errors, low-quality data).
Several factors contribute to a high degree of missingness. Sequencing errors, such as poor base calling, often result in incomplete data. Low-quality DNA samples, especially those extracted from degraded tissues, also increase the proportion of missing data. Experimental conditions, like the type of tissue or the genotyping batch, can further affect call accuracy. Studies have shown that DNA sourced from cell lines has a significantly higher MCR compared to blood samples (p<9 × 10−14). These issues highlight the importance of maintaining high-quality standards during data collection.
Why Does Missed Calls Rate Matter?
Impact on data quality and downstream analyses.
A high missed calls rate directly affects the quality of your dataset. Missing data can skew allele frequency calculations, leading to errors in identifying genetic associations. For instance, studies have found that the proportion of missing data correlates with experimental factors like tissue type and collection date. If left unchecked, these inconsistencies can compromise downstream analyses, such as genome-wide association studies (GWAS).
Consequences for research outcomes and decision-making.
Missing data can also influence research outcomes. A high degree of missingness may lead to biased results, reducing the reliability of your findings. For example, a study revealed that missing call rates were significantly higher in certain case categories (0.12% vs 0.10%, p<0.01). Such discrepancies can mislead decision-making, especially in clinical research. By addressing these issues, you can ensure that your dataset supports accurate and meaningful conclusions.
Identifying and Quantifying Missing Data

How to Calculate Proportion of Missing Data
Tools and methods for calculating missing data (e.g., PLINK, VCFtools).
To calculate proportion of missing data, you need specialized tools that analyze genetic datasets efficiently. PLINK and VCFtools are two widely used options. PLINK, a command-line tool, allows you to calculate proportion of missing data per site or per individual. For example, the command --missing in PLINK generates a report showing the missingness rate for each marker and sample. Similarly, VCFtools, designed for Variant Call Format (VCF) files, provides commands like --missing-site to calculate proportion of missing data per site. These tools simplify quantifying missingness, making it easier to identify problematic regions in your dataset.
Using these tools, you can pinpoint areas with high missingness and take corrective actions. For instance, if a specific marker shows a missing rate above 5%, you might exclude it from further analysis. This ensures your dataset remains reliable for downstream applications.
Setting thresholds for acceptable levels of missing data.
Setting thresholds for missing data is crucial for maintaining dataset quality. Most researchers consider a missing rate below 5% acceptable. However, this threshold may vary depending on your study's goals. For genome-wide association studies (GWAS), stricter thresholds like 1% are often applied to ensure robust results. Tools like PLINK and VCFtools allow you to filter data based on these thresholds. For example, you can use PLINK's --geno command to exclude markers exceeding a specified missing rate. By setting these limits, you ensure your dataset supports accurate and meaningful analyses.
Visualizing Missing Data
Heatmaps and other visualization techniques.
Visualizing missingness helps you understand patterns in your dataset. Heatmaps are a popular choice for this purpose. They display missing data as color-coded grids, making it easy to spot problematic regions. For instance, a heatmap might reveal that certain samples or markers have consistently high missingness. Tools like R's pheatmap package or Python's seaborn library can generate these visualizations. Another effective method involves bar plots, which summarize the proportion of missing data across samples or markers.
These visualizations provide actionable insights. If a heatmap shows clusters of missingness, you might investigate whether experimental conditions or sequencing errors caused the issue.
Interpreting patterns of missingness in datasets.
Patterns of missingness often reveal underlying problems in your dataset. For example, if missing data clusters around specific markers, it might indicate sequencing errors. Alternatively, high missingness in certain samples could result from poor DNA quality. Identifying these patterns helps you address root causes. For instance, you might re-sequence problematic regions or exclude low-quality samples. By interpreting these patterns, you can improve your dataset's reliability and ensure it meets the standards required for robust research.
Strategies to Handle Missing Data
Imputation Techniques for Missing Data
Overview of imputation methods (e.g., mean imputation, machine learning-based approaches).
Imputation is a powerful method for addressing missing data in genetic datasets. It involves estimating and filling in the gaps where data is incomplete. Common approaches include mean imputation, where missing values are replaced with the average of observed data. However, advanced methods like machine learning-based algorithms often yield better results. For example, Random Forest (RF) and k-Nearest Neighbors (kNN) have shown exceptional performance in genomic studies. RF is highly accurate but computationally intensive, while kNN offers a more practical solution for large datasets. These methods are particularly effective for non-coding variants, ensuring your dataset remains robust for downstream analyses.
Tools for imputation (e.g., BEAGLE, IMPUTE2).
Several tools simplify the imputation process. BEAGLE and IMPUTE2 are widely used for genetic datasets. BEAGLE excels in phasing and imputing genotypes, making it ideal for large-scale studies. IMPUTE2, on the other hand, is known for its accuracy in handling complex datasets. Both tools support VCF files, ensuring seamless integration with your existing data. By leveraging these tools, you can significantly reduce the proportion of missing data and enhance the reliability of your research.
Filtering Low-Quality Data
Removing low-quality samples or variants.
Low-quality samples or variants can inflate the rate of missing data. You should identify and remove these elements to maintain dataset integrity. For instance, samples with a high proportion of missing data or variants with low call rates often introduce biases. Tools like PLINK and VCFtools allow you to filter out these problematic entries efficiently. By doing so, you ensure that only high-quality data contributes to your analyses.
Setting quality control thresholds for inclusion.
Establishing quality control thresholds is essential for filtering data. Most researchers set a missing data threshold of 5% for markers and samples. For stricter studies, such as genome-wide association studies (GWAS), thresholds as low as 1% are common. Using tools like PLINK, you can apply these thresholds to your VCF files. For example, the --geno command in PLINK helps exclude markers exceeding the set limit. This practice ensures your dataset meets the standards required for accurate and meaningful results.
Balancing Data Quality and Quantity
Trade-offs between retaining data and ensuring accuracy.
Balancing data quality and quantity often involves trade-offs. Removing low-quality samples improves accuracy but reduces dataset size. Conversely, retaining more data may introduce errors. You must evaluate your study's goals to strike the right balance. For example, in studies with limited samples, you might tolerate slightly higher missing data rates to preserve statistical power.
Best practices for maintaining dataset integrity.
To maintain dataset integrity, follow best practices like regular quality checks and imputation. Use tools like BEAGLE or IMPUTE2 to address missing data in your VCF files. Visualize missingness patterns with heatmaps to identify problematic regions. Additionally, set clear thresholds for data inclusion and stick to them. These steps help you create a reliable dataset that supports robust research outcomes.
The Role of Sequence Depth in Reducing Missed Calls

Understanding Sequence Depth
Definition and importance of sequence depth in genetic datasets.
Sequence depth refers to the number of times a specific DNA segment is read during sequencing. Higher sequence depth increases the likelihood of accurately identifying genetic variants. For example, a depth of 30x means each base in the DNA sequence is read 30 times. This redundancy ensures that errors, such as false-positive or false-negative calls, are minimized. In genetic datasets, sequence depth directly impacts the reliability of variant calling, especially in regions with complex structures or low-quality DNA samples.
Adequate sequence depth is critical for reducing missed calls. Studies show that datasets with a depth below 10x often exhibit higher rates of missing data. By increasing depth, you can improve the accuracy of your dataset and reduce the need for extensive imputation or filtering.
How sequence depth affects call rates.
Sequence depth plays a pivotal role in determining call rates. Low depth often results in incomplete or ambiguous data, leading to missed calls. For instance, a depth of 5x might fail to detect rare variants, while a depth of 20x or more ensures better coverage and fewer gaps. Tools like VCFtools can help you analyze the relationship between depth and call rates in your dataset. By monitoring these metrics, you can identify areas requiring additional sequencing to enhance data quality.
Optimizing Sequence Depth
Strategies to ensure adequate sequence coverage.
To achieve adequate sequence coverage, you should plan your sequencing project carefully. Start by assessing the complexity of your target genome. For human genomes, a depth of 30x is often recommended. Use tools like PLINK or VCFtools to evaluate existing coverage and identify regions with low depth. If gaps are detected, consider targeted resequencing to fill them. Additionally, ensure that DNA samples are of high quality to maximize sequencing efficiency.
Another effective strategy involves pooling samples with similar characteristics. This approach reduces costs while maintaining sufficient depth for accurate variant calling. By combining these methods, you can ensure your dataset meets the required standards for robust analyses.
Balancing cost and data quality in sequencing projects.
Balancing cost and data quality is a common challenge in sequencing projects. High sequence depth improves call rates but increases expenses. To optimize both, prioritize regions critical to your study. For example, focus on coding regions or areas with known associations to your research question. Use cost-effective sequencing platforms and leverage tools like BEAGLE for imputation to address gaps without additional sequencing.
Sobot’s solutions can streamline this process by integrating data analysis tools into your workflow. With features like automated quality control and data visualization, you can monitor sequence depth and call rates efficiently. This ensures your project stays within budget while maintaining high data quality.
Actionable Steps to Reduce Missed Calls Rate with Sobot
Implementing Quality Control Pipelines
Key steps in a robust quality control process.
A robust quality control process ensures your genetic datasets remain reliable and accurate. Start by identifying problematic regions in your dataset using tools like PLINK or VCFtools. These tools help you detect high levels of missing data and low-quality variants. Next, set clear thresholds for data inclusion, such as a missing rate below 5%. Apply these thresholds consistently to maintain dataset integrity. Finally, use imputation tools like BEAGLE to address gaps in your data. This step minimizes the impact of missing data on downstream analyses.
Tools and software for automating quality control.
Automation simplifies quality control and reduces human error. Tools like PLINK and VCFtools automate tasks such as filtering low-quality variants and calculating missing data proportions. Sobot’s solutions integrate seamlessly with these tools, offering features like automated data visualization and quality checks. This integration saves time and ensures your datasets meet the highest standards.
Continuous Monitoring and Improvement
Regularly assessing dataset quality.
Regular assessments help you maintain high-quality datasets. Use visualization techniques like heatmaps to monitor patterns of missing data. Tools like VCFtools can generate reports on call rates and sequence depth, allowing you to identify areas needing improvement. Frequent evaluations ensure your data remains consistent and reliable.
Adapting strategies based on project needs and outcomes.
Your strategies should evolve with your project. For instance, if you notice high missing data rates in specific regions, consider resequencing those areas. Alternatively, adjust your quality control thresholds to balance data quality and quantity. Sobot’s tools provide real-time insights, helping you adapt quickly and effectively.
Leveraging Sobot's Expertise in Data Quality
How Sobot's solutions enhance data integrity and operational efficiency.
Sobot’s solutions streamline data management and improve operational efficiency. Features like automated quality control and intelligent routing ensure your datasets remain accurate. For example, Sobot’s tools can integrate with VCF files, automating tasks like filtering and imputation. This reduces manual effort and enhances data integrity.
Case study: Agilent's success in improving data quality with Sobot.
Agilent, a leader in life sciences, partnered with Sobot to address challenges in managing complex datasets. By using Sobot’s omnichannel workbench and AI-powered tools, Agilent achieved a 95% customer satisfaction rate and a sixfold increase in efficiency. The chart below highlights key performance metrics achieved through Sobot’s solutions:
| Metric | Value |
|---|---|
| Problem Resolution Rate | 85% |
| Customer Satisfaction Rate | 99% |
| Sign-off Rate Increase | 35% |
| COD Collection Rate Increase | 40% |
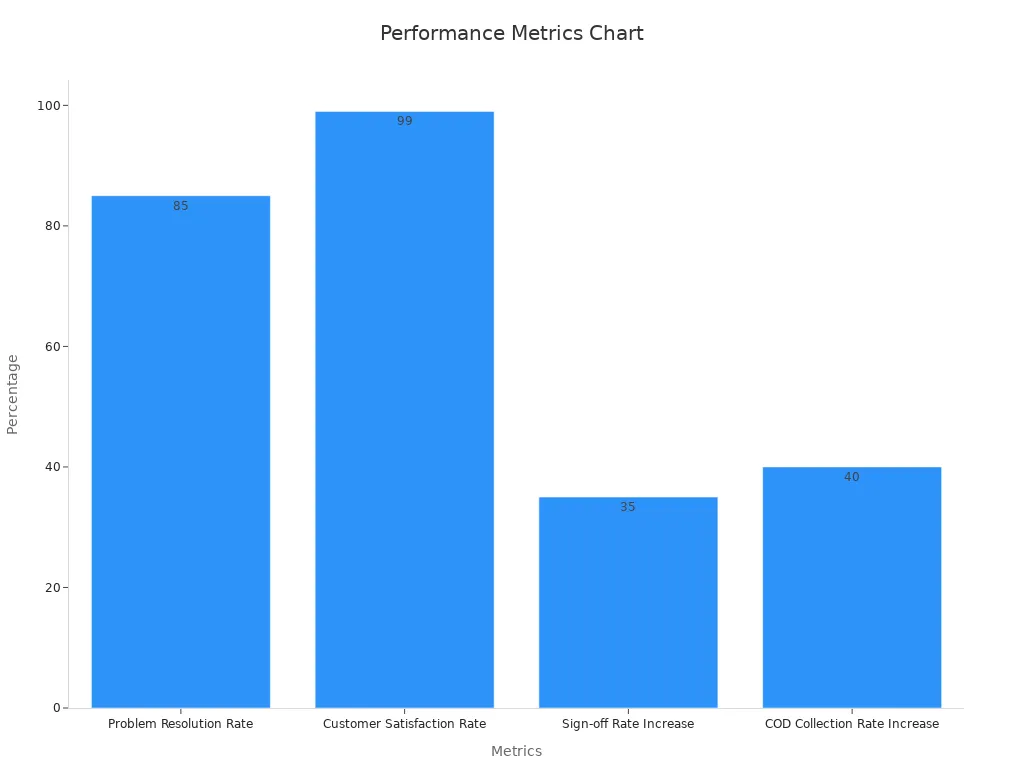
This collaboration demonstrates how Sobot’s expertise can transform data quality and operational outcomes.
Reducing missed calls rate (mcr) is essential for ensuring the accuracy of genetic datasets. High-quality data minimizes biases and improves research reliability. You can achieve this by implementing strategies like imputation, filtering low-quality data, and optimizing sequence depth. For example:
- Exclude individuals with high genotype failure rates to reduce false positives.
- Remove SNPs with excessive missing genotypes for better accuracy.
Best practices, such as using Random Forest or kNN for imputation, enhance dataset integrity. The table below highlights their effectiveness:
| Imputation Method | Effectiveness in Genomic Data |
|---|---|
| Random Forest | Best method for genomic data |
| kNN | More realistic approach |
Sobot’s tools, like automated quality control and VCF integration, simplify these processes. By adopting these solutions, you ensure reliable research outcomes.
FAQ
What is the acceptable threshold for missed calls rate in genetic datasets?
Most researchers consider a missed calls rate below 5% acceptable. For genome-wide association studies (GWAS), stricter thresholds like 1% are often used. These thresholds ensure reliable results by minimizing biases in downstream analyses. Tools like PLINK help you filter data based on these thresholds.
How can you visualize missing data in genetic datasets?
You can use heatmaps or bar plots to visualize missing data. Heatmaps highlight patterns of missingness, while bar plots summarize missing data proportions. Tools like R's pheatmap package or Python's seaborn library make it easy to create these visualizations.
What tools are best for imputing missing data?
BEAGLE and IMPUTE2 are widely used for imputing missing data in genetic datasets. BEAGLE excels in phasing and genotype imputation, while IMPUTE2 offers high accuracy for complex datasets. Both tools integrate seamlessly with VCF files, ensuring efficient data handling.
How does sequence depth affect missed calls rate?
Higher sequence depth reduces missed calls rate by improving the accuracy of variant identification. For example, a depth of 30x ensures reliable detection of genetic variants. Low depth, such as 5x, often leads to incomplete data and higher rates of missingness.
How does Sobot enhance data quality in genetic research?
Sobot's solutions automate quality control and data visualization, streamlining your workflow. For instance, Sobot integrates with tools like PLINK to filter low-quality data efficiently. This ensures your datasets meet high standards, improving research reliability and operational efficiency.
See Also
Best Call Center Analytics Tools to Use in 2024
Understanding The Efficiency Of Call Center Automation
Best Reviewed Automated Voice Calling Solutions for 2024
Enhancing Call Center Efficiency Through Effective Monitoring
